
Molecular graph representations and SELFIES: A 100% robust molecular string representation
SELFIES is a new molecular representation with an exciting set of properties and applications.

Mario Krenn, Alston Lo, AkshatKumar Nigam, Cynthia Shen, Luca Thiede, Seyone Chithrananda, and Alán Aspuru-Guzik
The goal of this post is to discuss SELFIES, a 100% robust molecular string representation, in the context of general molecular graph representations. One of our motivations is to get direct feedback from the community, suggestions and ideas for new useful extensions. First, we discuss in general how to represent molecules on a computer -- comparing string-based and adjacency-based (sometimes loosely called "graph-based") methods. After one simple example of SELFIES, we discuss several generative models enabled by its 100% robustness. Then, we briefly show two astonishing results that strongly indicate that SELFIES are easier for computers to "understand" than other representations. Finally, we state three very general open tasks for powerful string representations that should be tackled in the future.
For a technical discussion of what are SELIFES, see our paper in Machine Learning: Science & Applications and our extensive GitHub repo.
Thanks to numerous excellent suggestions from the community, the SELFIES language has developed significantly since its first introduction in the first preprint in mid-2019. For example, it can now handle all features that exist in SMILES, most of them in a semantically valid way. We want to actively encourage the community to continue to help us develop a robust string-based representation for molecules. Thus, we are highly interested in feedback from users, suggestions and ideas for future extensions, please post them as an issue/discussion at GitHub or send us an e-Mail: Mario & Alán. We plan to organize a workshop on molecular representations to elaborate on challenges and possibilities hopefully later this year.
Molecular representations in a computer
Artificial Intelligence (AI) technology has become a remarkable influence over the last few years. Its importance in science and technology is growing at an ever-increasing rate. The impact has left its marks also in cheminformatics and computational chemistry. There, AI technology augments humans' capabilities in solving challenging questions on designing novel functional molecules and advanced materials.
One central problem in this endeavour is the representation of molecules for computers. There are two main approaches, adjacency-matrix-based and string-based approaches to represent graphs. In the former, molecular graphs are represented by adjacency matrices where every element of the matrix indicates whether two vertices share a connection. This representation is sometimes loosely called "graph representation". The matrix needs to be augmented with a vector specifying the atom species of the individual vertices to use this representation for molecules. The simplicity of adjacency matrix representations, however, poses severe weaknesses. For instance, there is no natural way to represent 3-dimensional structures of molecules and many other chemical properties -- which are, of course, essential for the molecule's functionality.

Another way to represent molecules in the computer is strings. Strings are a much more powerful representation than adjacency matrices, as -- in principle -- strings can encode arbitrary computing programs. The standard method, SMILES, has been developed more than 30 years ago and has been a significant workhorse over the decades in chemoinformatics. SMILES can encode several molecules' spatial features and is sometimes called a 2.5-dimensional representation. However, now, in the era of machine learning in chemistry, SMILES face significant issues. The problems stem from the fact that SMILES strings have a complex grammar. When used in machine learning models, most of the results are entirely invalid. Either they do not correspond to correct syntactical molecules or violate fundamental constraints from physics and chemistry. Special-case solutions for specific generative models exist, but a universal solution needs to concern the representation itself.
The first significant effort to solve the issue of string-based representation for machine learning in chemistry has been DeepSMILES by Noel O'Boyle and Andrew Dalke. They redefined SMILES to circumvent several common syntactical errors. While DeepSMILES showed significant improvements over SMILES, not all syntactic mistakes were solved, and semantic mistakes (physical and chemical laws) were not addressed.
This is what we present: SELFIES, a 100% robust, simple, string-based molecular representation (GitHub). Even entirely random SELFIES strings represent correct molecular graphs. To do so, we exploit ideas from theoretical computer science, namely formal grammar and formal automatons. One can understand a SELFIES string as an elementary computer program that is transformed into a molecular graph with a simple compiler. The features of SELFIES: It is as powerful as SMILES (for instance, can represent 3d features of molecules), is human-readable (we come back to this in the end), and is easier for computers to "understand" (we explain below on several examples what we mean). We hope that SELFIES will help the community advance and develop new machine learning models that tackle some of the significant issues of the 21st century.

The basic idea of SMILES is to represent a chain of atoms. However, molecules are much richer than atom chains. Therefore, SMILES introduce two additional features to indicate branches and rings. Branches are represented as chains of atoms in brackets, that emerge from the main chain. Rings are represented by two numbers that indicate the atoms that share an additional edge.
SELFIES is a formal Chomsky type-2 grammar (or analogously, a finite state automata). This can be understood as a small computer program with minimal memory to achieve 100% robust derivation. It is designed with two ideas in mind: First, the non-local features in SMILES (rings and branches) are localized. Instead of indicating the beginning and end of a ring and branch in strings, SELFIES represents rings and branches by their length. After a ring and branch symbol, the subsequent symbol is interpreted as a number that stands for a length. This circumvents many syntactical issues with non-local features.
Second, physical constraints are encoded by different states of the deriving formal automaton/grammar. As an example, physically, a molecule of the form C=C=C is possible (three carbons connected via double bonds). However, F=O=F is not possible, because fluorine can only form one bond (not two) and oxygen can only form two bonds (not four as in this example). In SELFIES, after compiling a symbol into a part of the graph, the derivation state changes. This can be considered as a minimal memory that ensures the fulfilment of physical constraints.

The effects of these features can be clearly seen when we apply random mutations on the strings that describe MDMA (the example in the first image). We see that mutations are harmful to SMILES in most cases, while all mutated SELFIES correspond to valid molecular graphs. This indicates excellent advantages for generative models.
How to install and use SELFIES
SELFIES can easily be installed via pip:
pip install selfiesMany examples can be found at GitHub, and detailed documentation can be found here. In the following, I want to show an elementary example that translates between SMILES and SELFIES:
import selfies as sf benzene = "c1ccccc1" # SMILES --> SELFIES translation # '[C][=C][C][=C][C][=C][Ring1][Branch1_2]' encoded_selfies = sf.encoder(benzene) # SELFIES --> SMILES translation decoded_smiles = sf.decoder(encoded_selfies) # 'C1=CC=CC=C1 len_benzene = sf.len_selfies(encoded_selfies) # 8 symbols_benzene = list(sf.split_selfies(encoded_selfies)) # ['[C]', '[=C]', '[C]', '[=C]', '[C]', '[=C]', '[Ring1]', '[Branch1_2]'] SELFIES and de-novo design of molecules
Next, we show one main motivation of SELFIES: The application in de novo computational design of molecules. Here we show three conceptually different approaches that exploit the advantages of SELFIES.
How SELFIES enable advanced combinatorial approaches
How SELFIES can advance genetic algorithms
How SELFIES are used in three different deep generative models
1. STONED: An efficient combinatorial approach
After the development of SELFIES, a question often had us wondering: How powerful is a purely unbiased combinatorial, generative model with SELFIES that exploits random and systematic modifications of molecular strings? Attempting to solve the problem, we recently introduced the STONED algorithm (Superfast Traversal, Optimization, Novelty, Exploration and Discovery) that efficiently explores and interpolates in the chemical space. Surprisingly, we show that STONED can perfectly solve many commonly used cheminformatics benchmarks that before were thought to be challenging problems. Thereby, we demonstrate that the generation of diverse structures, rediscovery of molecules and other tasks are simple problems that can be solved purely combinatorial with SELFIES. We believe that STONED can be used for practical tasks where there can often be a lack of data and high-efficiency requirements. Important examples include the design of functional materials in material science and catalysis.

2. SELFIES in Genetic Algorithms
n a recent application, we demonstrated the use of SELFIES for an inverse design using genetic algorithms (GAs, see GitHub). Commonly, when GAs are developed using the SMILES molecular representation, sophisticated hand-crafted mutation rules and domain-knowledge have to be incorporated to guarantee molecular validity. With SELFIES, we can circumvent this effort and use arbitrary random modifications of molecular strings as the mutations in the evolutionary strategy. We apply the GA to commonly used benchmarks such as penalized logP, QED, and molecular similarity generation. Our architecture outperformed other generative models in efficiency and performance without incorporating domain knowledge. We can even augment our algorithm with neural networks that resemble some sense of chemical intuition. This allows for structure generation towards a known dataset while increasing molecular diversity. Shortly after, a group at Cyclica developed the algorithm Deriver, which also exploits SELFIES' robustness. They extended the ideas by using domain knowledge to improve the molecules' practical applicability.

4. SELFIES in a Deep Generative Models
Next, we show three different examples of how SELFIES advances or even enables several deep generative models. Those algorithms employ deep neural networks for the design of novel molecules. All of these models have in common that SELFIES generates syntactically and semantically valid molecules, thus no additional corrections or complex model architectures are required that are difficult to train. We will see how SELFIES’ 100% robustness can be exploited.
SELFIES in Variational Autoencoders
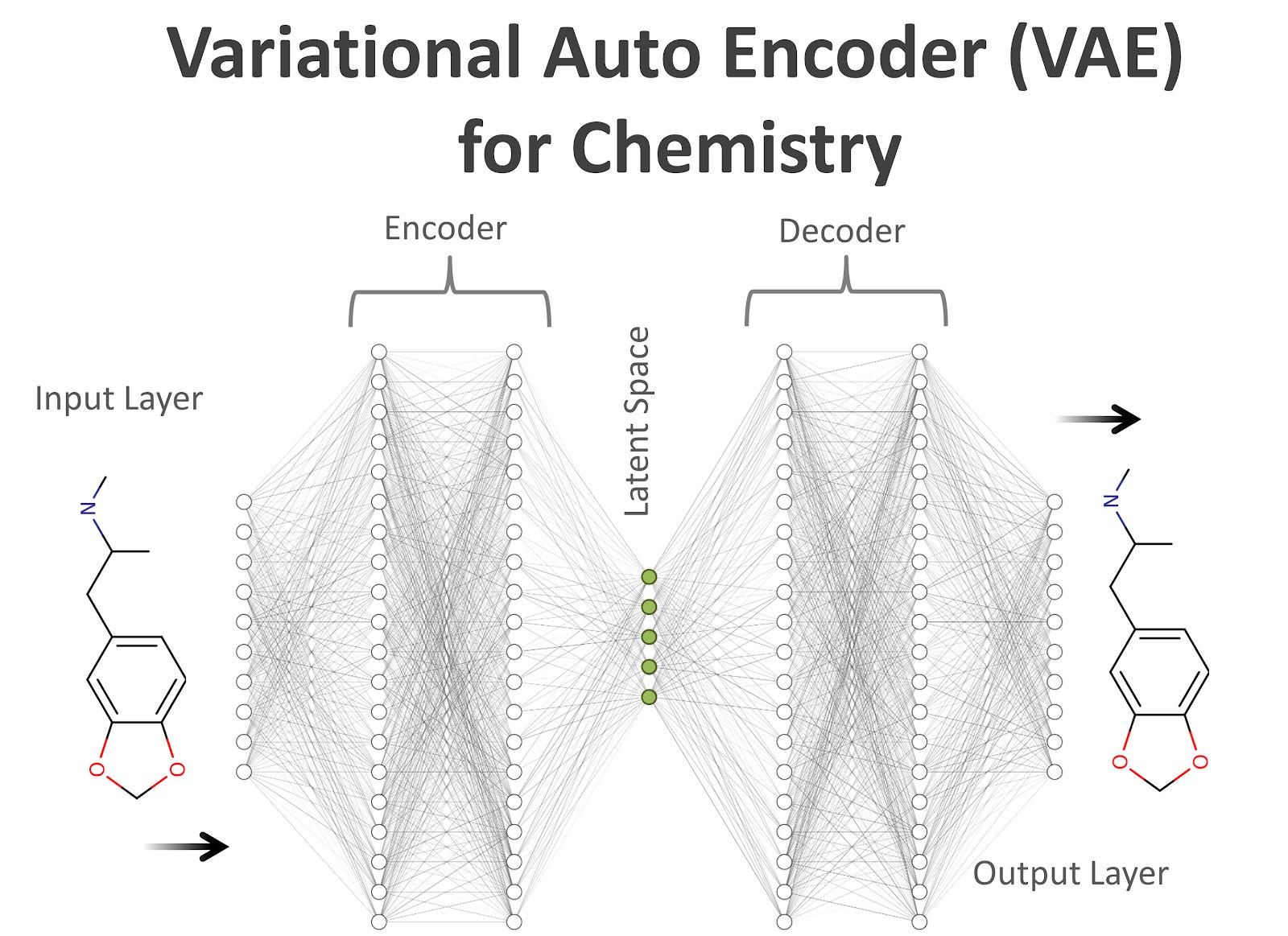
A variational autoencoder is a commonly-used generative model for molecular design. From discrete molecular graphs, the neural network creates a continuous internal latent representation of the molecular space. In this abstract space, we can subsequently optimize molecular properties.

Every point in the latent space corresponds to a molecule, transformed from the latent representation to the molecular representation (SMILES or SELFIES) with a decoding neural network. When sampling planes in this continuous vector space, we find that only small, unconnected regions correspond to valid molecular graphs. In contrast, every single point in the latent space is valid for SELFIES. This is a great advantage per se, and in the following, we show how this advantage can be exploited.

Of course, intuitively, if the entire latent space consists of valid molecular graphs, it’s diversity and density is larger. This is exactly what we observe. Sampling from the latent space until we cannot find new molecules shows that the SELFIES latent space encodes at least 2 orders of magnitude (!) more valid and potentially useful molecules than the SMILES and DeepSMILES latent spaces. The number of valid molecules can be considered the potential resource in molecular optimization; thus, SELFIES latent spaces show huge potential. It is important to recognize that we did not change the neural network model itself, just the molecular representation. That means, applying different clever machine learning allows us to further improve and extend the capabilities of the VAE, without being worried about whether the output makes physical sense. SELFIES guarantees that it does.
SELFIES and DeepDreaming

Another generative model, the Deep Molecular Dreaming algorithm PASITHEA, was enabled by SELFIES. PASITHEA is a gradient-based method inspired by the inceptionism methods in computer vision, which are used to observe the inner workings of deep neural networks, and thereby create dream-like images. The idea is to train a neural network to classify images into categories. After the classification quality is sufficiently high, the network weights are frozen, and the training is inverted: Now the goal is to generate a picture that optimizes a certain image class. As an example, we give the image a picture of chemical glassware and ask it to transform the image such that it looks more like an image of dogs.
Before, DeepDreaming has been used mainly for images and videos (quasi-continuous data structures) that allow continuous transformations. However, molecules are much more complex data structures, so at first, it is not clear how to apply DeepDreaming: A certain molecular string cannot continuously be transformed into another meaningful molecular string. However, if we use 1-hot encodings and allow for intermediate values between zeros and ones, we have introduced continuous transformations with values that can be optimized in a gradient-based way. If we would use SMILES, it is by no means obvious that our transformation will lead to a reasonable, chemically valid molecule. However, if the 1-hot encoding represents SELFIES strings, we can be sure of their validity. Thereby, DeepDreaming is generalized to the chemistry universe.
In addition to enabling inception as a design method in chemistry, SELFIES also enables the interpretation of the neural network's intermediate steps. In some sense, this allows the human to understand what neural networks learn about chemical properties it was trained on. Again, as all intermediate steps are valid molecules due to SELFIES, the human can extract new chemical rules by probing the network with suitable initial molecular strings. These rules, which are inherent to the neural network, may contribute to our own scientific understanding of the structure-property relationship.
SELFIES in Curiosity Driven Reinforcement Learning
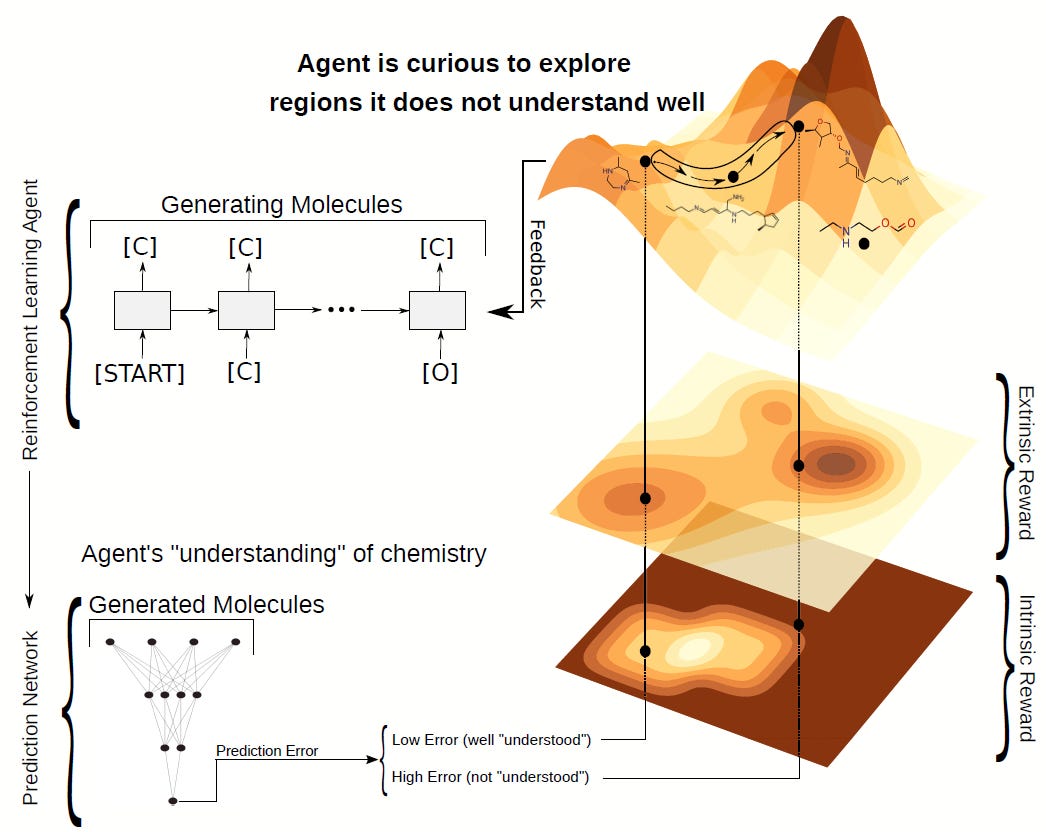
Reinforcement learning has gained a lot of attention in recent years for achieving superhuman performance on games like chess and Go. However, some authors also applied reinforcement learning to the physical sciences, among others, to generate new molecules. There, molecules are usually represented by SMILES strings, which require filtering many syntactically invalid strings, particularly at the beginning of training. Therefore, to learn the syntax of SMILES, previous attempts often pre-trained on a dataset. However, this takes time and biases the agent towards molecules of this specific data distribution.
In contrast, due to SELFIES being a 100% robust representation of molecules, the post-processing and pre-training can be omitted. SELFIES characters can just be used as actions in a plug and play fashion, and more complicated motives such as aromatic rings or functional groups can be added as extra actions without having to check if the actions are valid for a given string or not. This makes it much simpler to use, and the absence of pre-training opens the door to complete de novo training without any biases. In “Curiosity in exploring chemical space: Intrinsic rewards for deep molecular reinforcement learning” we explore precisely this possibility of bias-free molecular generation
SELFIES in Normalizing Flow Generative Models
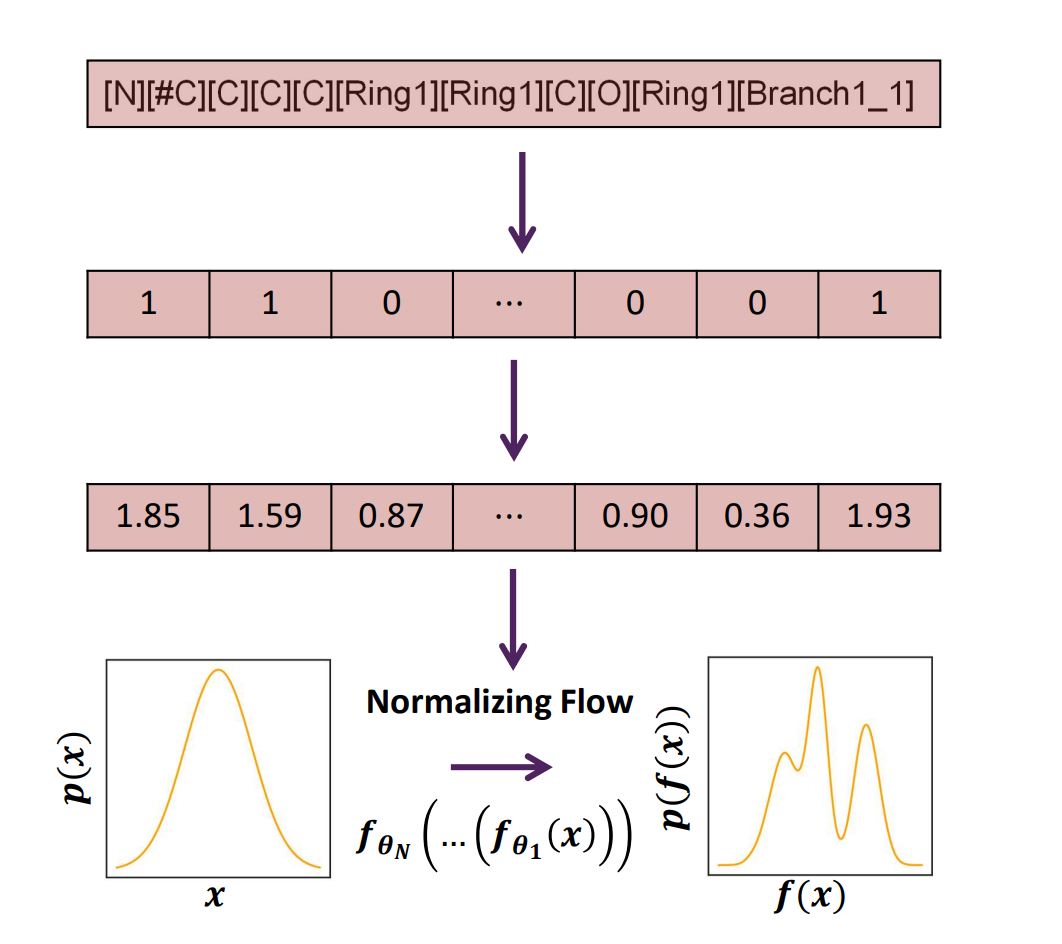
Recently, Nathan Frey and Bharath Ramsundar developed a molecular graph generation framework using normalizing-flow (NF) based models, SELF-Referencing Embedded Strings, and active learning. It generates a high percentage of novel, unique, and valid molecules and efficiently identifies optimal generated samples. Here, the authors do not take advantage of a compressed latent space (as for instance, in VAEs). Instead, the NF learns an invertible mapping between a simple base distribution and a target distribution. Using one-hot encodings of SELFIES representations as inputs to the normalizing flow, the architecture maps a simple base distribution to a complex target distribution.
Aiming to solve a common problem with molecular generative models - invalid outputs due to chemical rules not being encoded in the model architecture - they first encode the QM9 dataset as SELFIES strings. With an initial training set of only 200 small molecules in the QM9 dataset, 78% of the generated samples are chemically valid, not present in the training data, and unique simultaneously, which is a direct consequence of SELFIES’ robustness. With active learning, the maximally novel, drug-like generated samples are identified with an order of magnitude fewer steps than random search. The model is significantly simpler and easier to train than other molecular generative models and enables the fast generation and identification of novel drug-like molecules. Flow-based models accelerated with SELFIES offer a promising, low-data and compute-efficient strategy for molecular design.
SELFIES advances “machine-understandability”
The SELFIES representation has been developed to enable the applications and advances in generative models mentioned above. However, Kohulan Rajan, Achim Zielesny and Christoph Steinbeck at the University of Jena have made an incredible discovery. In two papers, they have shown strong indications that it is easier for machines to “understand” SELFIES (compared to other string-based representations such as SMILES and DeepSMILES).

Their first work has shown that machines can work best with SELFIES when they are tasked with translating an image of a molecule into a molecular string. This can be interpreted such that machine learning models find it easier to communicate chemistry using SELFIES.

In their follow-up work, they tasked the machine learning model in a different translation problem. This time, they wanted to get an IUPAC name from a molecular string. Again, they find that SELFIES outperformed other representations. For that reason, their STOUT algorithm has an inbuilt SMILES to SELFIES translator before giving the input to the neural network.
We find this surprising direction absolutely fascinating, and it will be exciting to answer questions of the sort “What makes the SELFIES language easier to learn for deep neural networks?”. This is not only important for applications but connects to important fundamental questions about machine understandability. For sure, more work in this direction will be much anticipated!
What is left to do with string representations?
We see three essential questions and tasks for the future for molecular representations.
1. New applications and domains benefiting from 100% robust representations
We wonder whether SELFIES will continue to outperform other representations in other, novel deep learning methods. Examples are the applications of Attention-based methods or Hopfield-Networks, for generative tasks. In general, string-based representations of Graphs are domain-independent. For example, graph-based tools developed for molecules can also be applied in quantum technology. It will be interesting to find more domains of applications, which allows for domain-independent machine-learning models -- one of the holy grails in AI.
2. Explore “machine-understandability” of representations
The results by Rajan, Zielesny and Steinbeck opened an entirely new research direction that deals with the question of which language is easiest to learn for deep neural networks. Given that this question is brand-new, much is unclear. As a first step, it will be exciting to understand why SELFIES outperforms other representations. This insight could be used to potentially advance the representation itself. As mentioned in the introduction, string representations can encode arbitrary computations (for instance, Turing machines). That means we have an enormous amount of freedom to explore. It would be surprising that we have found the best language with SELFIES already.
3. Explore “human-understandability” of representations
Arguments have been made that SMILES are easier to understand for humans than DeepSMILES or SELFIES. We do not fully agree. It might be easy to understand very small structures. Still, as soon as more intricate molecules with multiple rings and branches are investigated, humans have a hard time immediately “see” the corresponding molecular graphs. We wonder which of the existing string representations is most simple for humans to read. It would certainly be interesting to perform such empirical experiments. Finally, what are fundamentally the most simple representations, and how could it exploit the massive degrees of freedom provided by strings to be also strongly “machine-understandable”.
Many research questions remain open. In some sense, frankly, it seems we only scratched at the surface of possibilities.
Dear Dr. Alan! I really love this approach! I am PhD in Organic Chemistry and I have a second degree in Computer Science. I worked, many years ago, in a passionate project called SISTEMAT, where we developed a string definition of natural products. You article almost made me cry. Congratulations! @profjhgb
Thank you Joao for your extremely nice comments! I look forward to interacting in the future. I really appreciate your enthusiasm.